Harvard Medical School article featuring OGI researchers technique
The Gene Therapy Renaissance: How one experimental technique overcame a troubled legacy and today is helping the blind to see
R. ALAN LEO − April 18, 2013
In 1999, researchers at the University of Pennsylvania injected 19 people with a virus carrying a gene designed to correct a rare metabolic disease. Early results appeared promising: Among the first 17 adult subjects, the worst symptom was a fever, an expected response to the modified virus that carried the therapeutic gene.
The 18th subject was Jesse Gelsinger, who died.
Investigators still debate exactly what went wrong, but most agree that the delivery virus triggered a massive and fatal overreaction from the 18-year-old’s immune system. For the field of gene therapy, the Gelsinger case became shorthand for a sea of troubles.
Some saw a story of tragic hubris: a teenager paying with his life when researchers pushed too far, too fast. Others pointed to flaws in design and conduct specific to that trial, but the damage to the field was done. The Penn trial was halted immediately. So were other planned trials. Congressional hearings followed. Federal regulators and institutional review boards increased scrutiny that continues to this day. Investors shied away from new ventures and shelved ones already begun.
Gene therapy’s golden dawn had ended. But through the twilight years that followed, researchers learned from each setback and forged ahead. Today, many researchers investigating gene therapies and their biological underpinnings share an optimism long absent or long unseen. When it comes to conditions of the eye, gene therapists have achieved some of their most exciting successes to date. Researchers at HMS are building on that progress to help the blind to see.
Refocusing
One such researcher is Eric Pierce, the Solman and Libe Friedman Associate Professor of Ophthalmology at Harvard Medical School and director of the Ocular Genomics Institute at the Massachusetts Eye and Ear Infirmary.
But in 1999, the pediatric ophthalmologist and HST alum was just setting up his new academic lab at Penn’s Scheie Eye Institute, to investigate the genetics of retinal diseases. This was also the year Jesse Gelsinger died.
Though he wasn’t yet involved in gene therapy, Pierce was soon drawn to its potential after colleagues reported in 2001 that they had restored vision to a population of dogs with a condition nearly identical to a human retinal disease, Leber congenital amaurosis, or LCA. That team, led by husband-and-wife researchers Jean Bennett and Albert Maguire, injected the dogs’ retinas with a virus carrying a functional copy of a defective gene, RPE65.
Encouraged by those results, Pierce set up a retinal degeneration and genetics program in the Division of Ophthalmology at Children’s Hospital of Philadelphia, to prepare for the day if and when ocular gene therapy reached human trials. “I thought I was getting ahead,” Pierce recalls. “But within a year, Jean and Albert came to me and said, ‘We want to do a clinical trial. Would you help?’ so I just got started in time.”
That collaboration led to the most celebrated success in gene therapy. In 2008, the team reported that the first three patients, young adults, all saw modest improvement in retinal function with almost no adverse effects. Dozens have been treated since, with the greatest benefits accruing to the youngest patients.
“Between tests, kids would say, ‘I can ride my bicycle around the neighborhood’ or ‘I can play soccer on my own’—in other words, without an aide to help find the ball,” Pierce said. “Adults would say, ‘I can find my way to my seat in the restaurant for dinner, even if the lights are down’ or ‘I can see my kid play sports that I could never see before.’”
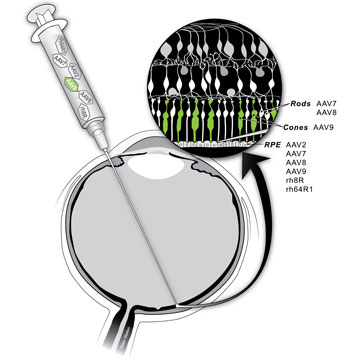
Setting sights
The LCA trial delivered a single gene—RPE65—to correct a single genetic defect. But the disease, which affects about one person in 80,000, is part of a much larger family of degenerative eye diseases known collectively as inherited retinal degenerations. The most common cause of inherited retinal degeneration, retinitis pigmentosa (RP) affects about one person in 3,000, and about half of those cases have been linked to mutations in any of 200 different disease genes. Researchers are working today to replicate the success of RPE65 therapy with some of these other genes—but they are also looking for ways to move beyond that scattershot approach.
“LCA is very specific. You’re replacing a diseased gene, but you can’t do that for every gene. That one trial may have cost $10 million, and you can’t do that for every disease,” said Joan Miller, Henry Willard Williams Professor of Ophthalmology and chair of the Department of Ophthalmology at HMS. “So you need to look for pathways that are common to classes of disease.”
That’s how Miller, who is chief of ophthalmology at Mass Eye and Ear, recruited Pierce. Her pitch: Build a center for collaboration among the world’s leading experts on retinal degenerations and other eye diseases to find common pathways and strategies to treat them.
Pierce joined the Berman-Gund Laboratory for the Study of Retinal Degenerations, whose director at the time, Eliot Berson, had built a large databank of 9,000 blood samples and 14,000 examination results from inherited retinal degeneration patients. Pierce directs the Ocular Genomics Institute, a group that includes glaucoma geneticist Janey Wiggs, Paul Austin Chandler Associate Professor of Ophthalmology, and HMS Professor of Neurology Elizabeth Engle, an expert on the genetics of eye movement disorders. “I don’t think there’s any other group around the world that’s got such a great combination of people focused on the genomics of eye disease,” Miller said. “The Ocular Genomics Institute is primed to do something big with genetics and gene-based therapies.”
Miller plucked another rising star from Penn, virologist Luk Vandenberghe. Vandenberghe’s specialty is adeno-associated viruses (AAVs), a promising class of “stealth” viruses that naturally infect humans without causing any known disease.
Revamping Vectors
Researchers point to the development of AAV vectors as a driving force in gene therapy’s comeback. Unlike the adenovirus given to Jesse Gelsinger, they don’t provoke the immune system while they deliver the desired gene. And unlike retroviruses used in other forms of gene therapy, they operate alongside, rather than within, the host’s DNA, avoiding potential cancer-causing mutations. (Their stealth comes with a price, however: a relatively small cap on the size of the genes they can carry.)
“AAV has chosen a path, it seems, to basically fly under the radar as a virus,” said Vandenberghe, now a member of the Ocular Genomics Institute and an HMS lecturer in ophthalmology at Schepens Eye Research Institute and Mass Eye and Ear. “And as a vector, that’s actually an ideal property because we do not want to alert the immune system to our presence, because that could lead to dire clinical consequences and the elimination of the genetic graft.”
This year, Vandenberghe opens the doors of a core facility to engineer new viral vectors and provide them to researchers across the HMS community. Among those researchers is Connie Cepko, Bullard Professor of Genetics and Neuroscience in the HMS Department of Genetics.
In the 1980s, Cepko had developed some of the first retroviral vectors as a postdoc at MIT and the Whitehead Institute. When she opened her own lab, she used some of these same tools to explore fundamental questions about how the eye develops. She kept an interest in gene therapy from the sidelines. Then came the first success of RPE65 therapy, in dogs.
“From that moment I knew that AAV would work, and I thought it would work in humans,” said Cepko, who is also a Howard Hughes Medical Institute Investigator. “So we started a new project to ask, ‘What could we do that would be a fairly generic gene therapy for the various genetic forms of blindness?’”
Common Vision
Cepko’s team began by asking: Why do photoreceptors die? The answer hinges on the two kinds of photoreceptors: rod photoreceptor cells, responsible for vision in dim light, and cone photoreceptor cells, responsible for color and detail. In most types of RP, the rods express a disease gene, while cones do not. This means that individuals with RP are born night-blind, but they can see quite well during the day—at first, anyway. Depending on the individual, color vision starts to fade during childhood, while in others it can fade as late as age 50.
“We thought if we could understand why the cones lose function and then die, there might be a generic way to combat that,” Cepko said. “Because no matter what the rod disease gene is, the cones always die.” They looked at four mouse models of RP, each with a different genetic defect in their rods, and asked what is common at the time cones start to die.
They found an answer in a metabolic regulator, mTOR, which plays a key role in cell growth, survival and proliferation. “We knew something was wrong with the cones very early on in the disease, because mTOR was not phosphorylated,” Cepko said. The researchers used insulin, which can trigger phosphorylation of mTOR, and found that this treatment prolonged the survival of the cones.
“So we said, okay, can we think of other ways to metabolically intervene and help these sick cones? We started thinking along these lines as daily injections with insulin over many years will not be a good therapy” Cepko said. “We have been asking, if there is a way we can augment the metabolism, sort of give the cells a booster, like a generic tonic for their metabolism?”
Now, Cepko is screening a handful of genes in mice that may provide that metabolic boost. Her lab develops some of its own viral vectors, and sources others from outside providers. Animal tests and vector development are time-consuming and profoundly expensive, she says—endeavors that would benefit from shared resources and facilities.
“Right now, every time we come up with an idea for a new gene to try, we run up against the expense of this type of work. The expenses limit what we do in a very serious way,” Cepko said. “A core here for basic science and preclinical work would be a fantastic resource and would help move things along.”
It’s a common vision among gene therapy researchers, and one Pierce and Vandenberghe would like to take a step further—a core facility certified to produce viral vectors for use not only in labs, but also in patients.
“Developing a clinical virus production facility in the Harvard community could facilitate many things,” Pierce said. “It could help us get to clinical trials faster. The intellectual propoerty of Harvard-derived vectors could be a revenue source. And the scale is such that having the larger Medical School community be part of that process could make it work better.”
The researchers are profoundly optimistic about the potential to treat not just one defect, but entire classes of eye disease, and other diseases as well.
It’s an optimism that Cepko contrasts with the blue-sky enthusiasm of the 1980s and ’90s, when the public—and some scientists—discussed gene therapy as a panacea for inherited disease.
“I think the original optimism was just not well founded in science,” Cepko said, “and I hold some of the scientists responsible for that. Richard Mulligan, one of the people who created this field, was right when he cautioned that the time course being touted was just too ambitious, considering where we were in our understanding of the vectors, their delivery, and the diseases.”
According to Cepko, this second wave of optimism is based on two things. First, researchers have learned a lot more about the technical aspects of the field, such as vector design, production, delivery, types of diseases to target, which vector for which disease, to name a few. And second, they have learned from the experiences in the clinic—including the disasters.
“The therapy in the eye didn’t have any disasters. It was just: AAV worked; we’re all very happy about that. It was an insightful choice for Bennett and her colleagues to use AAV, and their choice was based upon good science. But it’s also good fortune. Many things might not have worked. Future attempts might fail due to some technical issues, but it is important that this first case did not have safety issues. It will spur the development of gene therapy for many other diseases. We have come to appreciate how idiosyncratic each case will be, and thus not all will work. But having such a wonderfully successful trial in the eye gives us great encouragement, and it is very likely that at least some other attempts also will be successful.”